Theme 1
Controlling the Early Stages of Crystallisation in Droplets
Many of the most important features of a crystal – such as its structure or orientation on a substrate – are defined at nucleation. Learning how to direct nucleation processes, such that we can select one polymorph over another, or induce nucleation when and where we want, is therefore one of the ultimate challenges in crystallisation. We are exploiting the unique capabilities of droplet systems to achieve this goal, and are investigating how different additives direct nucleation, and gain control by deploying them at different points along the reaction pathway. Crystallisation is being characterised in droplets in our lab and at synchrotron beam lines. This combines techniques that can be used to probe crystallisation while the droplets are still on-chip with complementary methods that allow us to extract the droplets to characterise these processes ex situ. Our PMMA and Kaptan devices allow us to study crystallisation in droplets at a synchrotron using SAXS, WAXS, and EXAFS. We are also developing methods to siphon off the droplets at different time-points so that they can be further characterised.
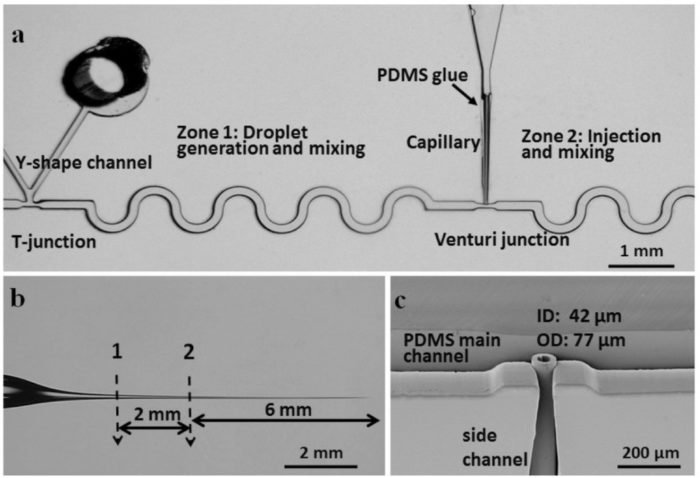
Droplet Microfluidic Chip with Passive Pico-injector
(https://doi.org/10.1002/smll.201702154)
1.1 Droplet Microfluidics XRD Identifies Effective Nucleating Agents for Calcium Carbonate
The ability to control crystallisation reactions is required in a vast range of processes including the production of functional inorganic materials and pharmaceuticals, and the prevention of scale. However, it is currently limited by a lack of understanding of the mechanisms underlying crystal nucleation and growth. To address this challenge it is necessary to carry out crystallisation reactions in well-defined environments, and ideally to perform in situ measurements. Here, a versatile technique is presented that uses a microfluidic device and synchrotron radiation to meet these demands. Droplet Microfluidics-Coupled X-ray Diffraction (DMC-XRD) enables the collection of time-resolved, serial diffraction patterns from a stream of flowing droplets containing growing crystals. The droplets offer reproducible reaction environments, and radiation damage is effectively eliminated by the short residence time of each droplet in the beam. DMC-XRD is then used to identify effective particulate nucleating agents for calcium carbonate, and to study their influence on the crystallisation pathway. Bioactive glasses and NX illite are shown to significantly lower the induction time, highlighting the importance of both surface chemistry and topography on the nucleating efficiency of a surface. This technology is also extremely versatile, and could be used to study dynamic reactions with a wide range of synchrotron-based techniques.
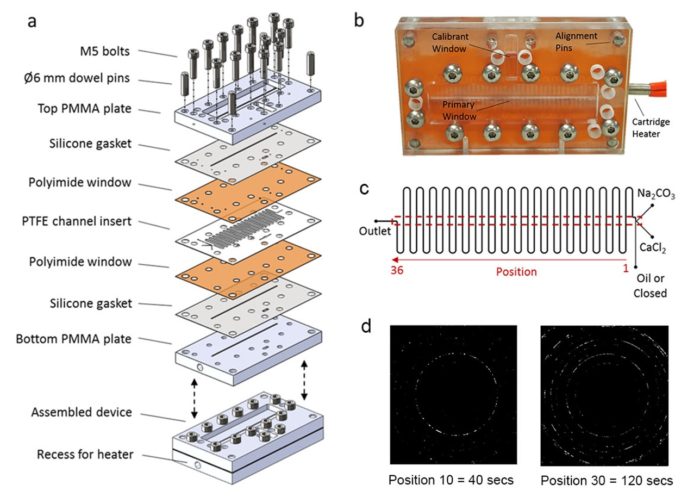
Microfluidic device for studying crystallization in flowing droplets in situ using X-ray based techniques. (a) Construction, (b) appearance of final device, (c) schematic of flow system and (d) XRD images of CaCO3 precipitation recorded at different times.
(https://doi.org/10.1002/adfm.201808172)
1.2 Coupling Droplet Microfluidics and X-rays Scattering: Innovative Data Processing to Investigate Nanoparticle Synthesis
X-ray scattering techniques provide a powerful means of characterizing the formation of nanoparticles in solution. Coupling these to segmented-flow microfluidic devices that offer well-defined environments gives access to in situ time-resolved analysis, excellent reproducibility and eliminates potential radiation damage. However, analysis of the data-sets generated can be extremely time-consuming, where these comprise frames corresponding to the droplets, the continuous phase and the interface between them. We here describe a robust, low-cost and versatile droplet microfluidics device and use it to study the formation of magnetite nanoparticles with simultaneous synchrotron SAXS and WAXS. Lateral outlet capillaries facilitate the X-ray analysis and reaction times of between a few seconds and minutes can be accommodated. A novel data processing method is then described that exploits the unique WAXS signatures of the droplets, continuous phase and interface region to identify the frames corresponding to the droplets. These are sorted and the background scattering is subtracted using an automated frame-to-frame approach, allowing the signal from the nanoparticles to be isolated from the raw data. Modeling these data gives quantitative information about the evolution of the sizes and structures of the nanoparticles. This versatile platform can be readily employed to study a wide range of dynamic processes in heterogeneous systems.
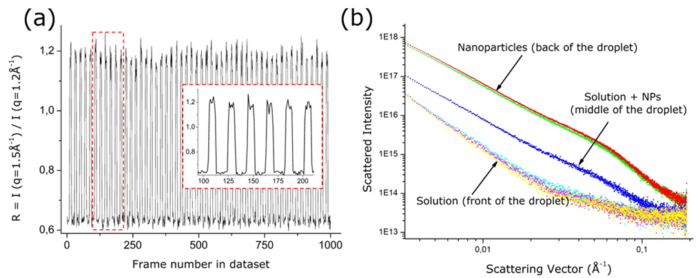
(a) Evolution of the selection criterion as a function of the frame number for an entire dataset derived from flowing droplets. The magnified inset demonstrates the stability of the droplet generation and the robustness of this parameter. (b) Consecutive SAXS frames within a dataset of flowing droplets. Three kinds of frames are obtained corresponding to solution only, nanoparticles-rich volume, or a low density of nanoparticles.
1.3 Dynamic Crystallization Pathways of Polymorphic Pharmaceuticals Revealed in Segmented Flow with Inline Powder XRD
Understanding the transitions between polymorphs is essential in the development of strategies for man-ufacturing and maximizing the efficiency of pharmaceuticals. However, this can be extremely challenging: crystallization can be influenced by subtle changes in environment such as temperature and mixing inten-sity or even imperfections in the crystallizer walls. Here, we highlight the importance of in situ measure-ments in understanding crystallization mechanisms, where a segmented flow crystallizer was used to study the crystallization of the pharmaceuticals urea:barbituric acid (UBA) and carbamazepine (CBZ). The reac-tor provides highly reproducible reaction conditions, while in situ synchrotron powder X-ray diffraction (PXRD) enables us to monitor the evolution of this system. UBA has two polymorphs of almost equivalent free-energy and so is typically obtained as a polymorphic mixture. In situ PXRD uncovered a progression of polymorphs from UBA III to the thermodynamic polymorph UBA I, where different positions along the length of the tubular flow crystallizer correspond to different reaction times. Addition of UBA I seed crys-tals modified this pathway such that only UBA I was observed throughout, while transformation from UBA III into UBA I still occurred in the presence of UBA III seeds. Information regarding the mixing-dependent kinetics of the CBZ form II to III transformation was also uncovered in a series of seeded and unseeded flow crystallization runs, despite atypical habit expression. These results illustrate the importance of cou-pling controlled reaction environments with in situ XRD to study the phase relationships in polymorphic materials.
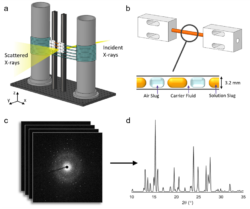
(a) Diagram of the analysis module of the KRAIC-D operating in transmission mode with X-ray penetration initiated behind the window section. (b) Enlarged view of a single analysis window comprising a Kapton tube and two polytetrafluoroethylene (PTFE) unions (inset shows the triphasic solution, air, and carrier fluid slug flow). (c) Accumulation of 2D diffraction pattern frames from 100ms exposures that are combined to achieve a (d) 1D diffraction plot (shown here is a combined scan of carbamazepine form II).
(https://doi.org/10.1021/acs.analchem.0c00860)
1.4 Evaluation of Microflow Configurations for Scale Inhibition and Serial X-ray Diffraction Analysis of Crystallization Processes
The clean and reproducible conditions provided by microfluidic devices are ideal sample environments for in situ analyses of chemical and biochemical reactions and assembly processes. However, the small size of microchannels makes investigating the crystallization of poorly soluble materials on-chip challenging due to crystal nucleation and growth that result in channel fouling and blockage. Here, we demonstrate a reusable insert-based microfluidic platform for serial X-ray diffraction analysis and examine scale formation in response to continuous and segmented flow configurations across a range of temperatures. Under continuous flow, scale formation on the reactor walls begins almost immediately on mixing of the crystallizing species, which over time results in occlusion of the channel. Depletion of ions at the start of the channel results in reduced crystallization towards the end of the channel. Conversely, segmented flow can control crystallization, so it occurs entirely within the droplet. Consequently, the spatial location within the channel represents a temporal point in the crystallization process. Whilst each method can provide useful crystallographic information, time-resolved information is lost when reactor fouling occurs and changes the solution conditions with time. The flow within a single device can be manipulated to give a broad range of information addressing surface interaction or solution crystallization.
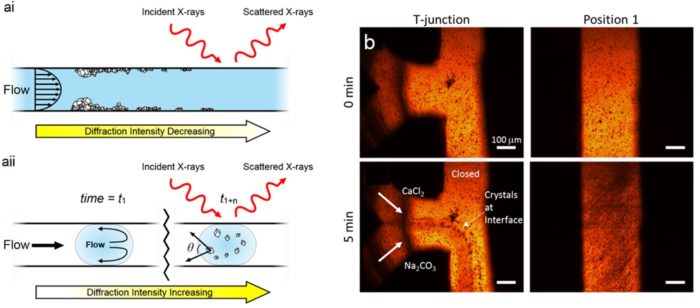
(a) Diagrams of microfluidic X-ray analysis of crystallization in (i) continuous and (ii) segmented flow. (b) Optical micrographs taken from the ambient temperature continuous flow experiment at the indicated time and position.
(https://doi.org/10.1039/D0LC00239A)